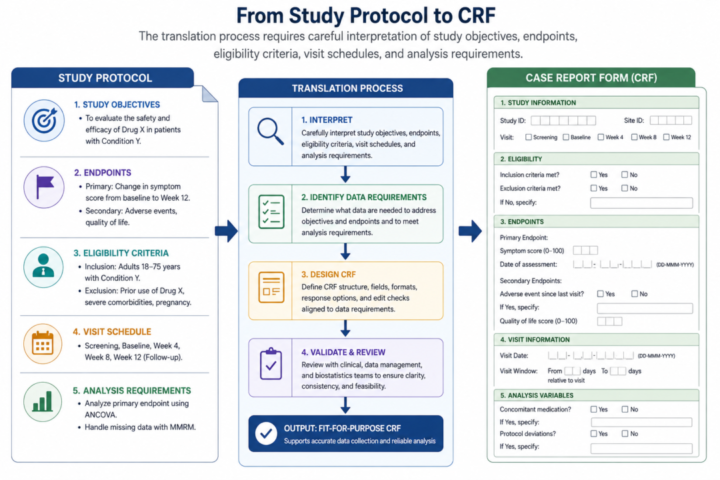
The study protocol is the central scientific, operational, and regulatory document for a clinical research study. It describes why the study is being conducted, what question it intends to answer, who will be included, what procedures will be performed, how participant safety will be protected, what outcomes will be measured, and how the data will be analyzed. For the clinical data manager, the protocol is more than a narrative description of a study. It is the blueprint from
which data requirements, case report forms, database structures, validation checks, monitoring reports, and analysis datasets are derived.
Every well-designed database begins with careful reading of the protocol. This reading is different from a general scientific review. The data manager reads with a particular set of questions in mind. What are the study objectives? What variables are needed to answer each objective? Which data are collected once, and which are collected repeatedly? Which procedures occur at screening, enrollment, follow-up, close-out, or unscheduled visits? Which values are needed for eligibility determination? Which values are needed for safety reporting?
Which values will appear in the final statistical analysis plan? Which values require source verification? Which fields contain confidential identifiers? Which fields should be coded rather than entered as free text?
The protocol usually contains several sections that directly inform data design. The background and rationale explain the clinical or public health problem and may reveal important context for variable selection. The objectives and hypotheses define the scientific questions.
The study design section explains whether the study is a trial, cohort, case-control study, cross sectional study, registry, or surveillance system. Eligibility criteria define inclusion and exclusion data. The schedule of events specifies when each procedure occurs. The safety section describes adverse event capture and reporting. The statistical analysis section describes outcomes, covariates, subgroup variables, and analytical populations.
In practice, protocols vary in quality and completeness. Some protocols provide very detailed schedules and outcome definitions, while others leave operational details unclear. A data manager must be able to identify ambiguities early and raise them with investigators before CRFs or databases are built. For example, a protocol may state that “fever resolution will be assessed at follow-up,” but may not specify whether fever resolution is based on measured temperature, caregiver report, absence of symptoms, or clinician judgment. If this ambiguity is not resolved before CRF design, different sites may collect the outcome differently, making analysis unreliable.
Protocol translation also requires attention to feasibility. A protocol may request variables that are scientifically interesting but difficult to collect in the intended setting. In a rural facility, a laboratory result may not be available on the same day. In a busy emergency department, lengthy forms may disrupt care. In a multisite study, some hospitals may use electronic health records while others rely on paper files. A good data manager does not simply build every requested field. They help the study team examine whether each field is necessary, measurable,
and collectable with acceptable quality.